May 26, 2025
PART 1: Who Gets It And Why

What this article is about: This article explains Alzheimer’s disease (AD): its development in the brain, causes, risk factors (especially the APOE ϵ4 gene), and ways to potentially reduce risk through lifestyle. We also preview future research directions and diagnostic advances. For the sake of your brain, the article is in two parts. Part 1 is a deep dive into the prevalence, pathophysiology, risk factors and diagnosis of AD. Part 2 is a deep dive into risk reduction and future research.
Why it’s important for you: Given its increasing impact, understanding AD is crucial. This article offers key insights into its complexities, potential for action, and the evolving landscape of diagnosis and treatment.
Key Takeaways:
- AD progresses from memory issues to significant cognitive decline.
- The APOE ϵ4 gene is a major risk factor, affecting brain processes.
- Newer blood tests offer less invasive ways to detect AD pathology.
- Brain insulin resistance is a significant factor in AD.
- Early attention to sleep, movement, nutrition, cholesterol, and metabolic health may reduce risk.
- Future AD research is actively pursuing targeted therapies.
PART 1: Who Gets It And Why
Read Time: About 12 minutes
We’re all mortal
Have you read Atul Gawande’s book, “Being Mortal”? It’s one of the most harrowing books I’ve ever read. In fact, I can’t even claim to have completed it – it was all too much for me after getting through half of it.
The book discusses issues of human mortality and the slow decline that many suffer in the latter decades of their lives. It has shaped thinking about the purpose of families and my approach as a doctor when called to nursing homes (originally invented as facilities to undergo a stint of rehabilitation, as opposed to living out the rest of your dying days!).

I consider my work in nursing homes (and hospital residency) my national service. I was repeatedly asked to assess patients with dementia so severe that they barely resembled people. Incoherent. Confused. Incontinent. And that characteristic stare our subconscious interprets instantly – the lights are on, yet nobody’s home. Like the staff, I’d fight the overwhelming urge to get in and get the hell out. I mean, the patient’s own family hadn’t visited for 3 months, so why should I hang around for longer than absolutely necessary in their depressing, piss-stinking cubicle, flipping through cut-and-pasted nurses’ notes and pages-long drug charts. Oh, I see, you don’t know what decade it is, but they’ve got you on something for cholesterol?!
All the things we value as humans: autonomy, freedom, variety, choice, open spaces, connection, conversation, intimacy, privacy and dignity are the diametrical opposite of how nursing home residents live. This is why I frequently remind my 4 kids, there’ll be no nursing home for me (unless, of course, you want to trigger a clause that sends my entire estate to the Hawthorn Football Club). I can’t say I’ve worked out the details, but if I forget who you are, you better get me a big dose of heroin and send me out with a bang! This article is about Alzheimer’s Disease (AD), a condition that slowly robs people of what makes them them and, not surprisingly, is the leading cause of nursing home admissions. It has a strong hereditary component and has no cure. Despite this miserable combination, even the most stoic among us are better off understanding the risks, mitigating factors, testing and treatments, and current research into the condition.

It is hard to talk about Alzheimer’s without first understanding what it does.
Alzheimer’s disease (AD) is a neurodegenerative disorder¹, characterised by progression from subtle memory impairment to profound cognitive deterioration and functional dependence. It often presents as episodic memory deficits, particularly for recent events, while semantic memory and procedural skills remain relatively preserved. As the disease advances, language difficulties², visuospatial deficits, executive dysfunction³, and behavioural changes emerge. Late-stage disease⁴ is marked by global cognitive impairment, loss of independence in activities of daily living, and neuropsychiatric symptoms⁵ including agitation, apathy, and psychosis.
The risk of developing AD increases significantly with age. For people aged 65 and over, about 1 in 14 (7%) are affected, rising to 1 in 6 (17%) for those aged 80 and older. After the age of 85, the incidence is 33%⁶.
The disease disproportionately affects women, who account for two-thirds of cases. Survival after diagnosis is usually 4 to 8 years⁷.
The biological correlate of AD⁸ is the pathological accumulation of amyloid-beta plaques and tau neurofibrillary tangles in the brain⁹. These changes begin 15-20 years before symptom onset, creating a substantial preclinical phase where intervention may be effective¹⁰.
One gene, Apolipoprotein E (ApoE), that is strongly associated with the condition, though the 2020 Lancet Commission found that up to 40% of dementia cases are linked to modifiable risk factors¹¹. These will be outlined later in this article.
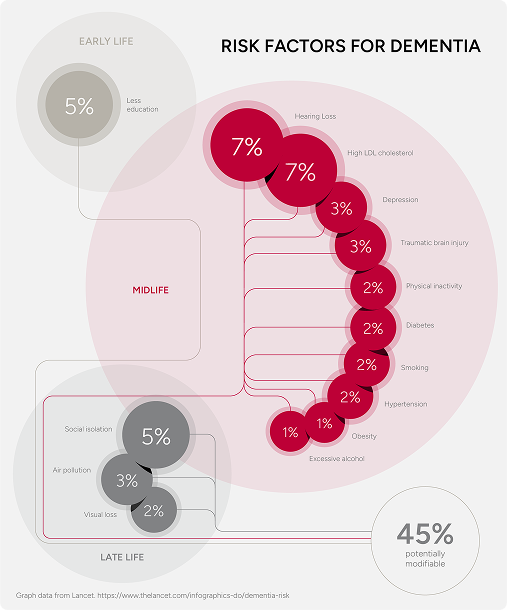
Up to 40% of dementia cases are linked to modifiable risk factors¹¹.
Is ApoE Our Best Lead Yet?
Apolipoprotein E (APOE) is the best-established genetic risk factor for AD. Meta-analyses suggest it accounts for approximately 25% of attributable genetic risk¹². Three primary variants (alleles) exist: ε2, ε3 and ε4. The ε3 allele is the most common in around 78% of the population¹³. Below, a table of risk by genotype is presented.
The presence of one ε4 (referred to as a “carrier”) increases the risk of AD by up to four times¹⁴. Two copies may raise the risk by up to twelve times¹⁵.
Longitudinal cohort studies with decades of follow-up show that some people with two ε4 alleles never develop AD¹⁶. While others, with no ε4 at all, still do. This variability underscores that AD is neither necessary nor sufficient to occur.
Figure 2. How the ApoE ε4 Allele Impacts AD Risk
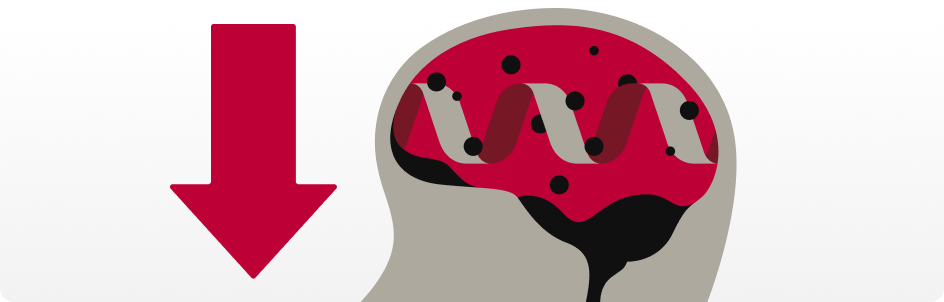
Waste Accumulation
Those with the ε4 Allele don’t clear amyloid beta as efficiently¹⁷, allowing it to accumulate¹⁸.

Inflammation
In ε4 carriers, microglia — the brain’s resident “cleanup” cells — adopt a persistently reactive state. Rather than quietly removing waste, they release higher levels of pro-inflammatory signals (like IL-1β and TNF-α), which can damage synapses and impair neuron function¹⁹. This heightened state of alert also makes it harder for the brain to clear amyloid-beta, fueling the fire²⁰.
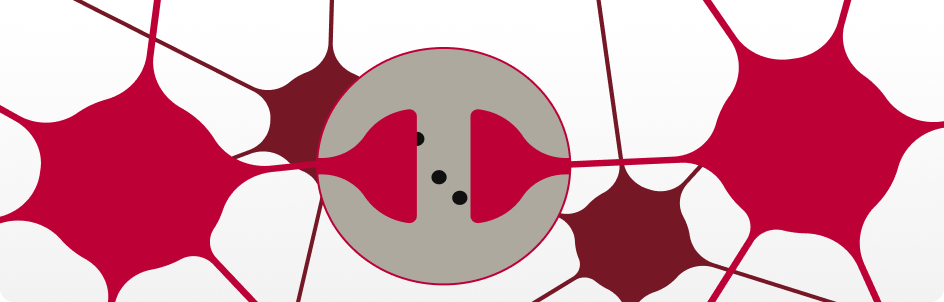
Altered Cellular Communication
The system that helps neurons (brain cells) talk to each other starts to break down due to disrupted fat (lipid) transport²¹.

Compromised Barrier
The blood-brain barrier, which normally shields the brain, becomes less effective, making the brain more vulnerable²².
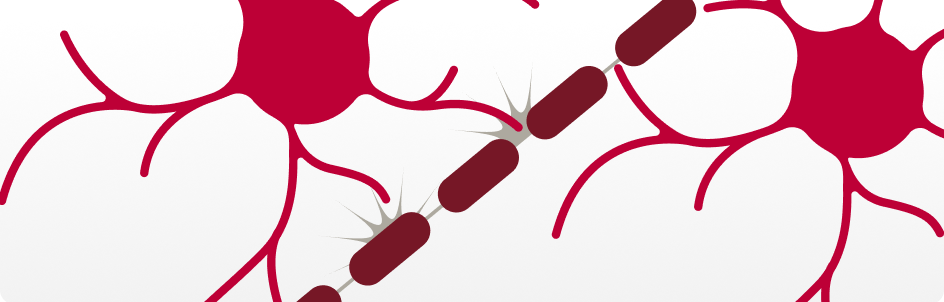
Reduced Energy Production
Astrocytes — cells that help power neurons — don’t produce energy as well, leaving brain cells with less fuel²³.
Early Signs Before Damage Appears
These energy issues begin long before visible brain damage appears on scans²⁴.
Systemic Effects
People with the ε4 gene often have higher insulin resistance (so more insulin is required to store nutrients), leading to more background inflammation and issues processing fats effectively²⁵ ²⁶.
Vicious Cycle
Systemic issues such as chronic inflammation, insulin resistance, and disrupted fat metabolism affect physical health and intensify brain changes, creating a self-reinforcing cycle that accelerates cognitive decline²⁷ ²⁸.
The APOE ε4 gene increases the likelihood of developing Alzheimer’s disease in both men and women. Individuals with one copy of the gene face roughly a threefold higher risk than non-carriers. This effect is seen in both sexes, though marginally stronger in women.²⁹ Notably, between the ages of 65 and 75 — a period when incidence begins to accelerate — female carriers are nearly six times more likely to develop Alzheimer’s than women without the gene. For men, the increase during this window is just over threefold. The gene appears to act more aggressively in women during this critical decade³⁰.
Progression follows suit. Among those with mild cognitive impairment, female ε4 carriers are 2.16 times more likely to develop AD than non-carriers; for men, the risk rises by 1.64. These hazard ratios reflect the relative risk of developing AD over time³¹.
Table 1. ApoE Genotype Risk Stratification
GENOTYPE
ε3 / ε3
POPULATION
60%
LIFETIME RISK
10% – 15%
MEDIAN AGE OF ONSET
84.3 years old
ODDS OF REACHING 85 WITHOUT AD
80% – 85%
GENOTYPE
ε2 / ε3
POPULATION
11%
LIFETIME RISK
6.2%
MEDIAN AGE OF ONSET
90.8 years old
ODDS OF REACHING 85 WITHOUT AD
88% – 92%
GENOTYPE
ε2 / ε2
POPULATION
1%
LIFETIME RISK
3.1%
MEDIAN AGE OF ONSET
93.5 years old
ODDS OF REACHING 85 WITHOUT AD
>95%
GENOTYPE
ε3 / ε4
POPULATION
23%
LIFETIME RISK
23% – 30%
MEDIAN AGE OF ONSET
75.5 years old
ODDS OF REACHING 85 WITHOUT AD
65% – 70%
GENOTYPE
ε2 / ε4
POPULATION
2%
LIFETIME RISK
19.8%
MEDIAN AGE OF ONSET
78.4 years old
ODDS OF REACHING 85 WITHOUT AD
72% – 77%
GENOTYPE
ε4 / ε4
POPULATION
2% – 3%
LIFETIME RISK
51% – 68%
MEDIAN AGE OF ONSET
68.4 years old
ODDS OF REACHING 85 WITHOUT AD
32% – 38%
This table details how different ApoE allele combinations influence Alzheimer’s risk across six dimensions: population frequency, lifetime risk, hazard ratio (relative to ε3/ε3), median age of onset, and likelihood of reaching age 85 without the disease. The presence of the ε4 allele sharply increases both relative and absolute risk, lowers the expected age of symptom onset, and reduces the odds of cognitive longevity. In contrast, ε2 is associated with delayed onset and greater resilience.
Advances in testing, from PET scans to blood tests, are ushering in a new era of Alzheimer’s diagnosis.
Quantitative positron emission tomography (PET) enables visualisation of early damage well before memory declines, detecting reduced cerebral glucose uptake, especially in critical regions (e.g. posterior cingulate, precuneus, and temporoparietal cortices). These patterns confirm that metabolic dysfunction is an initiating factor in cognitive decline³².
ε4 carriers often show greater sensitivity to metabolic stressors, including poor glycaemic control, disrupted sleep, and chronic inflammation. This reinforces the importance of early interventions addressing these factors³³ ³⁴ ³⁵.
Until recently, confirming AD pathology meant relying on cerebrospinal fluid analysis or PET imaging — both accurate but costly, invasive, and often unavailable outside major centres.
A newer option, the PrecivityAD blood test, offers a simpler, less invasive way to assess AD pathology.
It measures two proteins linked to the disease process — amyloid-beta and phosphorylated tau — and combines them into a single index known as the Amyloid Probability Score 2 (APS2)³⁶. This estimates the likelihood that amyloid plaques are present in the brain.
Precivity AD2 is reported to detect amyloid-PET positivity with 88% sensitivity and 89% specificity³⁶, meaning it identifies disease when present and rarely makes a call of disease when it isn’t actually there.
A separate real-world study involving more than 1,200 symptomatic patients found the test’s negative predictive value ranged from 87% to 92%, confirming its utility in reliably ruling out AD pathology, even in lower-prevalence primary care settings³⁷.
Current guidance restricts use to adults aged 55 and over who already show signs of cognitive impairment³⁸. It is not approved for screening asymptomatic individuals. Within its intended scope, however, PrecivityAD2 offers a non-invasive, analytically rigorous alternative to PET or CSF, expanding access to diagnostic certainty without compromising accuracy.
Is AD Actually ‘Type 3 Diabetes’?
Evidence suggests similarities between AD and other insulin resistance syndromes. Indeed, AD is increasingly referred to as ‘Type 3 Diabetes’.³⁹
People with Type 2 diabetes have a significantly increased risk of developing AD, and the risk rises in line with insulin resistance (assessed with a glucose tolerance or HOMA-IR blood test), even if diabetes hasn’t been diagnosed⁴⁰.
Insulin resistance in the brain initiates a cascade of pathological events associated with AD⁴¹. This includes increased production of neurotoxic amyloid-beta, impaired clearance of protein waste, and modification to another key protein called tau. Notably, these processes precede macroscopic changes detectable by standard brain imaging. Studies reveal that even in cognitively healthy individuals, cerebral insulin resistance correlates with regional brain atrophy in areas vulnerable to AD⁴². Furthermore, reduced cerebral glucose metabolism is observed years, even decades, before the onset of clinical symptoms. In APOE ϵ4 carriers, imaging suggests that metabolic decline may occur before amyloid accumulation, highlighting brain metabolism as a critical target for preventing cognitive decline⁴³.
Medical Disclaimer
The information provided by My Performance Doctor on this website, including written content, videos, and other materials, is intended solely for general informational and educational purposes. It does not constitute medical advice, diagnosis, or treatment and should not be relied upon as a substitute for professional medical judgment.
Using this site or its content does not create a doctor-patient relationship. Any application of the information provided is at the user’s own discretion and risk.
You should not delay or disregard seeking medical advice based on anything you read or view here. Always consult your healthcare provider regarding any medical concerns, symptoms, or conditions.

ABOUT THE AUTHOR
For over 30 years, Dr. Weisinger has been dedicated to improving health, performance, and well-being. His expertise spans clinical care, research, and academic instruction. Discover why so many trust Dr. Weisinger for personalised, proactive healthcare—because true health is about thriving, not just surviving.